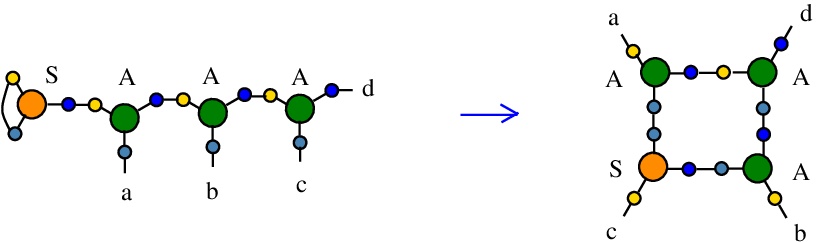
As Large Language Models are both random and asemantic, it seems that asemantic computing wins the world.
In this post I group some quotes from famous people, collected during early days (2013-2014) when “asemantic computing” was called simply “no semantics“.
First please do not forget to see what “asemantic computing” means, by looking either at the newest:
(doi) (figshare) M. Buliga, Argument from AI summary: How does asemantic computing differ from traditional distributed computing? figshare. Journal contribution. (2023)
or at:
M. Buliga, Asemantic computing, in: chemlambda. (2022). chemlambda/molecular: Molecular computers which are based on graph rewriting systems like chemlambda, chemSKI or Interaction Combinators (v1.0.0). Zenodo.
Now, quotes:
Rodney Brooks, Intelligence without representation (1987), (link to pdf) (saved pdf). Section 5.1. [Mentioned in the Nothing vague in the non semantic point of view. ]
Brooks cites as reference [10] the following:
M.L. Minsky, ed., Semantic Information Processing (MIT Press, Cambridge, MA, 1968)
Brooks quote:
“It is only the observer of the Creature who imputes a central representation or central control. The Creature itself has none; it is a collection of competing behaviors. Out of the local chaos of their interactions there emerges, in the eye of an observer, a coherent pattern of behavior. There is no central purposeful locus of control. Minsky
[10] M.L. Minsky, ed., Semantic Information Processing (MIT Press, Cambridge, MA, 1968)
gives a similar account of how human behavior is generated. […]
… we are not claiming that chaos is a necessary ingredient of intelligent behavior. Indeed, we advocate careful engineering of all the interactions within the system. […]
We do claim however, that there need be no explicit representation of either the world or the intentions of the system to generate intelligent behaviors for a Creature. Without such explicit representations, and when viewed locally, the interactions may indeed seem chaotic and without purpose.
I claim there is more than this, however. Even at a local level we do not have traditional AI representations. We never use tokens which have any semantics that can be attached to them. The best that can be said in our implementation is that one number is passed from a process to another. But it is only by looking at the state of both the first and second processes that that number can be given any interpretation at all. An extremist might say that we really do have representations, but that they are just implicit. With an appropriate mapping of the complete system and its state to another domain, we could define a representation that these numbers and topological connections between processes somehow encode.
However we are not happy with calling such things a representation. They differ from standard representations in too many ways. There are no variables (e.g. see
[1] P.E. Agre and D. Chapman, Unpublished memo, MIT
Artificial Intelligence Laboratory, Cambridge, MA (1986)
[Agre mentioned here at Phil Agre’s orbiculus]
for a more thorough treatment of this) that need instantiation in reasoning processes. There are no rules which need to be selected through pattern matching. There are no choices to be made. To a large extent the state of the world determines the action of the Creature. Simon
[14] H.A. Simon, The Sciences of the Artificial (MIT Press,
Cambridge, MA, 1969)
noted that the complexity of behavior of a system was not necessarily inherent in the complexity of the creature, but Perhaps in the complexity of the environment. He made this analysis in his description of an Ant wandering the beach, but ignored its implications in the next paragraph when he talked about humans. We hypothesize (following Agre and Chapman) that much of even human level activity is similarly a reflection of the world through very simple mechanisms without detailed representations.”
V. Braitenberg, Vehicles, Experiments in synthetic psychology, MIT Press (1986) (archive.org link) (saved pdf) From the end of Vehicles 3 section:
“But, you will say, this is ridiculous: knowledge implies a flow of information from the environment into a living being ar at least into something like a living being. There was no such transmission of information here. We were just playing with sensors, motors and connections: the properties that happened to emerge may look like knowledge but really are not. We should be careful with such words.”
Kappers, A.M.L.; Koenderink, J.J.; Doorn, A.J. van, Local Operations: The Embodiment of Geometry, Basic Research Series (1992), pp. 1 – 23 (link to pdf) (saved pdf)
[Mentioned in The front end visual system performs like a distributed GLC computation]
Quotes from the section 1, indexed by me with (a), … (e):
- (a) the front end is a “machine” in the sense of a syntactical transformer (or “signal processor”)
- (b) there is no semantics (reference to the environment of the agent). The front end merely processes structure
- (c) the front end is precategorical, thus – in a way – the front end does not compute anything
- (d) the front end operates in a bottom up fashion. Top down commands based upon semantical interpretations are not considered to be part of the front end proper
- (e) the front end is a deterministic machine […] all output depends causally on the (total) input from the immediate past.
Louis Kauffman amusing answer to such quotes (taken from Nothing vague in the non semantic point of view )
“
Dear Marius,
It is interesting that some people (yourself it would seem) get comfort from the thought that there is no central pattern.
I think that we might ask Cookie and Parabel about this.
Cookie and Parabel and sentient text strings, always coming in and out of nothing at all.
Well guys what do you think about the statement of MInsky?
Cookie. Well this is an interesting text string. It asserts that there is no central locus of control. I can assert the same thing! In fact I have just done so in these strings of mine.
the strings themselves are just adjacencies of little possible distinctions, and only “add up” under the work of an observer.
Parabel. But Cookie, who or what is this observer?
Cookie. Oh you taught me all about that Parabel. The observer is imaginary, just a reference for our text strings so that things work out grammatically. The observer is a fill-in.
We make all these otherwise empty references.
Parabel. I am not satisfied with that. Are you saying that all this texture of strings of text is occurring without any observation? No interpreter, no observer?
Cookie. Just us Parabel and we are not observers, we are text strings. We are just concatenations of little distinctions falling into possible patterns that could be interpreted by an observer if there were such an entity as an observer?
Parabel. Are you saying that we observe ourselves without there being an observer? Are you saying that there is observation without observation?
Cookie. Sure. We are just these strings. Any notion that we can actually read or observe is just a literary fantasy.
Parabel. You mean that while there may be an illusion of a ‘reader of this page’ it can be seen that the ‘reader’ is just more text string, more construction from nothing?
Cookie. Exactly. The reader is an illusion and we are illusory as well.
Parabel. I am not!
Cookie. Precisely, you are not!
Parabel. This goes too far. I think that Minsky is saying that observers can observe, yes. But they do not have control.
Cookie. Observers seem to have a little control. They can look here or here or here …
Parabel. Yes, but no ultimate control. An observer is just a kind of reference that points to its own processes. This sentence observes itself.
Cookie. So you say that observation is just self-reference occurring in the text strings?
Parabel. That is all it amounts to. Of course the illusion is generated by a peculiar distinction that occurs where part of the text string is divided away and named the “observer” and “it” seems to be ‘reading’ the other part of the text. The part that reads often has a complex description that makes it ‘look’ like it is not just another text string.
Cookie. Even text strings is just a way of putting it. We are expressions in imaginary distinctions emanated from nothing at all and returning to nothing at all. We are what distinctions would be if there could be distinctions.
Parabel. Well that says very little.
Cookie. Actually there is very little to say.
Parabel. I don’t get this ‘local chaos’ stuff. Minsky is just talking about the inchoate realm before distinctions are drawn.
Cookie. lakfdjl
Parabel. Are you becoming inchoate?
Cookie. &Y*
Parabel. Y
Cookie.
Parabel.
Best,
Lou”
This is somehow premonitory, how about a discussion today, involving Cookie, Parabel and a LLM?