… along with a short discussion about what the chemical concrete machine can compute. I start with this and then go to the example.
Until now I proved that the graph rewriting system called “chemical concrete machine” can do combinatory logic. Therefore it can compute anything (a Turing machine can). I shall be more specific, because to the eyes of somebody who is not used with functional programming, this might seem outrageous.
It can compute anything which can be computed by using any programming language based on lambda calculus, like Haskell or Scala. And then, some more (as regards only the things those languages can do without using any syntactic sugar, of course). The procedure is straightforward, namely translate the (essentially) combinatory logic terms into graphs and then let the enzymes (moves) to do their magic. I shall give a bit later the example of the if-then-else structure.
There are, of course, interesting questions to ask, among them:
- do exist real molecules and real enzymes which react one with another like in the chemical concrete machine formalism? Here I need your help, dear chemists.
- is this an example of a sequential or parallel computation?
- what evaluation procedure is used in the chemical concrete machine?
The second question is the most easy to ask: is parallel computation. The molecules (graphs) are mixed in a reactor with a choice of enzymes and then all possible reactions can occur in parallel, at all times. (Realistic models will have attached a probabilistic part, but there is nothing special here, because the chemical concrete machine, initialized with molecules and enzymes, is just a chemical reaction network, so any procedure to attach the probabilistic part to a chemical reaction network can also be used in conjunction with the chemical concrete machine.)
But we can also prepare the initial molecules such that the computation is sequential. It suffices to use zippers. More later. But for the moment, it is worthy to mention that the chemical concrete machine (as well as the graphic lambda calculus) are already proven to be more powerful than lambda calculus. Why? for at least two reasons:
- lambda calculus, or combinatory logic, are just sectors of the formalisms, i.e. they correspond to only a part of what the formalisms can do. There are other sectors, as well, for example the tangle diagram sector.
- they are naturally parallel, as long as we use only local moves, as is the case for the chemical concrete machine, for example. Indeed, I wrote that a graph is a “molecule”, but this is only a way of speaking, because molecules could be better identified with connected graphs. But in these formalisms the graphs are not supposed to be connected: any (finite) collection of graphs (i.e. molecules) is also a graph. The moves being local, there is no interference appearing in the simultaneous application of several instances of the (local) moves in different places, for different molecules (connected subgraphs) or even for the same molecule, as long as the places where the moves are applied are different one from another. On the other side, lambda calculus and combinatory logic are naturally sequential.
The third question, concerning the evaluation procedure will be also explored in further posts. Care has to be taken here because there are no variables in these formalisms (which translates in less demand of different species of real molecules only for the need to name variables). So it is about the order of moves, right? The short answer is that it depends, sometimes the computation done by the chemical machine can be seen as greedy evaluation, sometimes as lazy evaluation.
Let me make again the point that somehow the chemical concrete machine formalism should be seen as part of the beautiful idea of algorithmic chemistry. So, it’s not so unearthly.
Finally, it is well known that lambda calculus and Turing machines are the two pillars of computation. For historical reasons chemists seem to concentrate only on the emulation of Turing machines (pleas correct me if I’m wrong). The main idea of algorithmic chemistry, as far as I understand, is that a sufficiently complex chemical network has the capacity to do lambda calculus. But, if you are determined to use only Turing machines for chemical computing, then, supposing that algorithmic chemistry idea is true, you have to translate the natural language of lambda calculus into the Turing machine frame. This is a tarpit. Very fast, it becomes very hard to follow. Instead, why not use lambda calculus as it is, for the real powerful applications of chemical computing, and in parallel use one of the excellent languages for simulating in silico chemical computing.
_________________________
The if-then-else construct has already been explored in graphic lambda calculus, see Teaser: B-type neural networks in graphic lambda calculus (II) in . Here I shall do a much simpler example, just by making the right translations, as explained in the beginning of this post.
In lambda calculus, there are terms called TRUE, FALSE and IFTHENELSE, which are the Church encodings of the booleans true, false and if-then-else. Theassociated graphs in the chemical concrete machine are:

Take other two molecules A, B, with one exit each, in particular you may think that they correspond to terms in lambda calculus (but it is not mandatory). Then IFTHENELSE TRUE A B should become A. In the chemical concrete machine, with only beta + enzymes, look what is happening:

Along this chain of reactions, there is no other choice than the one from the figure. Why? Because essentially at every step there is only one reaction site available to the enzyme beta+ (of course, in the region of the reactor represented in the figure). The result is, unsurprisingly, compatible with the lambda calculus version, with the exception that A and B are not supposed to be (graphs corresponding to) lambda terms. They can be anything, as for example, from the family of “other molecules”.
In lambda calculus IFTHENELSE FALSE A B should become (by reductions) B. In the chemical concrete machine look what happens:

The previous remarks apply here as well.
With a little bit of imagination, if we look closer to what TRUE and FALSE are doing, then we can adapt the IFTHENELSE to what I’ve called a B-type NN synapse and obtain a molecule which releases, under the detection of a certain molecule, the medicine A, and under the detection of another the medicine B.
_____________________
 which are associated to terms in lambda calculus by
which are associated to terms in lambda calculus by 
 … or by letters
… or by letters  …. (what’s in a name?). These decorations are called “types”. There is only one operation, called “
…. (what’s in a name?). These decorations are called “types”. There is only one operation, called “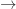 “, which takes a pair of types
“, which takes a pair of types  and returns the type
and returns the type  .
. gate. (We are not going to use it in relation to the lambda calculus sector.) As you see, this is compatible with the trivial quandle with the operation
gate. (We are not going to use it in relation to the lambda calculus sector.) As you see, this is compatible with the trivial quandle with the operation  .
. combinator.
combinator.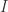 combinator (the identity) and for the
combinator (the identity) and for the 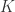 combinator.
combinator.
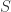 combinator:
combinator:

 .
.












 , if you apply a FAN-OUT gate to
, if you apply a FAN-OUT gate to  then you obtain two
then you obtain two 
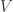 , the subbundle
, the subbundle  and the metric
and the metric 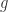 on
on 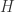 ) which were involved in the construction of the CC metric.
) which were involved in the construction of the CC metric.
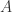 and
and  are molecules from the formalism of the chemical concrete machine and
are molecules from the formalism of the chemical concrete machine and 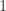 and
and 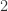 are labels. The blue arrow means any chemical reaction which is allowed.
are labels. The blue arrow means any chemical reaction which is allowed.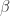 zippers, introduced in
zippers, introduced in 

 and
and  . Remark that:
. Remark that:

 , with
, with 
 enzymes, there is only one reaction site available, namely the one involving the red and green nodes in the neighbourhood of the
enzymes, there is only one reaction site available, namely the one involving the red and green nodes in the neighbourhood of the  . So there is only one reaction possible with a
. So there is only one reaction possible with a  and a new, shorter
and a new, shorter  , so the reaction with the enzyme
, so the reaction with the enzyme 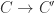 and a new, shorter zipper. The reaction continues like this, freeing in order the molecules
and a new, shorter zipper. The reaction continues like this, freeing in order the molecules  , then
, then 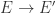 .
.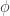 zipper). It behaves the same as the previous one, but this time in the presence of the FAN-IN enzyme
zipper). It behaves the same as the previous one, but this time in the presence of the FAN-IN enzyme 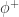 .
.



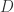 and
and  with
with 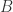 and
and  . Now, when we add further the
. Now, when we add further the 
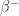 for the second reaction, and moreover a reaction site, denoted by closed red, dashed, curve. The meaning is that the enzyme react with the molecule by binding at the reaction site and by modifying it, without any other action on the rest of the molecule.
for the second reaction, and moreover a reaction site, denoted by closed red, dashed, curve. The meaning is that the enzyme react with the molecule by binding at the reaction site and by modifying it, without any other action on the rest of the molecule.






 of unspecified molecules, which can react (or not) one with another, this is left to the choice of the user of the chemical concrete machine. Besides this bag of names of molecules, we have a short list of essential molecules, which are the ingredients of the machine.
of unspecified molecules, which can react (or not) one with another, this is left to the choice of the user of the chemical concrete machine. Besides this bag of names of molecules, we have a short list of essential molecules, which are the ingredients of the machine.


 . Also functional programming, as far as I understand, also use this eta reduction, being more interested into types.
. Also functional programming, as far as I understand, also use this eta reduction, being more interested into types.