This is the 5th (continuing from part I and part II and part III and part IV) in a series of expository posts where we put together in one place the pieces from various places about:
- how is treated lambda calculus in chemlambda
- how it works, with special emphasis on the fixed point combinator.
I hope to make this presentation self-contained. (However, look up this page, there are links to online tutorials, as well as already many posts on the general subjects, which you may discover either by clicking on the tag cloud at left, or by searching by keywords in this open notebook.)
_________________________________________________________
This series of posts may be used as a longer, more detailed version of sections
- The chemlambda formalism
- Chemlambda and lambda calculus
- Propagators, distributors, multipliers and guns
- The Y combinator and self-multiplication
from the article M. Buliga, L.H. Kauffman, Chemlambda, universality and self-multiplication, arXiv:1403.8046 [cs.AI], which is accepted in the ALIFE 14 conference, 7/30 to 8/2 – 2014 – Javits Center / SUNY Global Center – New York, (go see the presentation of Louis Kauffman if you are near the event.) Here is a link to the published article, free, at MIT Press.
_________________________________________________________
2. Lambda calculus terms as seen in chemlambda continued.
Let’s look at the structure of a molecule coming from the process of translation of a lambda term described in part IV.
Then I shall make some comments which should be obvious after the fact, but useful later when we shall discuss about the relation between the graphic beta move (i.e. the beta rule for g-patterns) and the beta reduction and evaluation strategies.
That will be a central point in the exposition, it is very important to understand it!
So, a molecule (i.e. a pattern with the free ports names erased, see part II for the denominations) which represents a lambda term looks like this:

In light blue is the part of the molecule which is essentially the syntactic tree of the lambda term. The only peculiarity is in the orientation of the arrows of lambda nodes.
Practically this part of the molecule is a tree, which has as nodes the lambda and application ones, but not fanouts, nor fanins.
The arrows are directed towards the up side of the figure. There is no need to draw it like this, i.e. there is no global rule for the edges orientations, contrary to the ZX calculus, where the edges orientations are deduced from from the global down-to-up orientation.
We see a lambda node figured, which is part of the syntactic tree. It has the right.out port connecting to the rest of the syntactic tree and the left.out port connecting to the yellow part of the figure.
The yellow part of the figure is a FOTREE (fanout tree). There might be many FOTREEs, in the figure appears only one. By looking at the algorithm of conversion of a lambda term into a g-pattern, we notice that in the g-patterns which represent lambda terms the FOTREEs may appear in two places:
- with the root connected to the left.out port of a lambda node, as in the g-pattern which correspond to Lx.(xx)
- with the root connected to the middle.out port of an Arrow which has the middle.in port free (i.e. the port variable of the middle.in of that Arrow appears only once in that g-pattern), for example for the term (xx)(Ly.(yx))
As a consequence of this observation, here are two configurations of nodes which NEVER appear in a molecule which represents a lambda term:

Notice that these two patterns are EXACTLY those which appear as the LEFT side of the moves DIST! More about this later.
Remark also the position of the the insertion points of the FOTREE which comes out of the left.out port of the figured lambda node: the out ports of the FOTREE connect with the syntactic tree somewhere lower than where the lambda node is. This is typical for molecules which represent lambda terms. For example the following molecule, which can be described as the g-pattern L[a,b,c] A[c,b,d]

(but with the port variables deleted) cannot appear in a molecule which corresponds to a lambda term.
Let’s go back to the first image and continue with “TERMINATION NODE (1)”. Recall that termination nodes are used to cap the left.out port of a lambda lode which corresponds to a term Lx.A with x not occurring in A.
Finally, “FREE IN PORTS (2)” represents free in ports which correspond to the free variables of the lambda term. As observed earlier, but not figured in the picture, we MAY have free in ports as ones of a FANOUT tree.
I collect here some obvious, in retrospect, facts:
- there are no other variables in a g-pattern of a lambda term than the port variables. However, every port variable appears at most twice. In the graphic version (i.e. as graphs) the port variables which appear twice are replaced by edges of the graph, therefore the bound lambda calculus variables disappear in chemlambda.
- moreover, the free port in variables, which correspond to the free variables of the lambda term, appear only once. Their multiple occurrences in the lambda term are replaced by FOTREEs. All in all, this means that there are no values at all in chemlambda.
- … As a consequence, the nodes application and lambda abstraction are not gates. That means that even if the arrows of the pattern graphs appear in the grammar version as (twice repeating) port variables, the chemlambda formalism has no equational side: there are no equations needed between the port variables. Indeed, no such equation appears in the definition of g-patterns, nor in the definition of the graph rewrites.
- In the particular case of g-patterns coming from lambda terms it is possible to attach equations to the nodes application, lambda abstraction and fanout, exactly because of the particular form that such g-patterns have. But this is not possible, in a coherent way (i.e. such that the equations attached to the nodes have a global solution) for all molecules!
- after we pass from lambda terms to chemlambda molecules, we are going to use the graph rewrites, which don’t use any equations attached to the nodes, nor gives any meaning to the port variables other than that they serve to connect nodes. Thus the chemlambda molecules are not flowcharts. There is nothing going through arrows and nodes. Hence the “molecule” image proposed for chemlambda graphs, with nodes as atoms and arrows as bonds.
- the FOTREEs appear in some positions in a molecule which represents a lambda term, but not in any position possible. That’s more proof that not any molecule represents a lambda term.
- in particular the patterns which appear at LEFT in the DIST graph rewrites don’t occur in molecules which represent lambda terms. Therefore one can’t apply DIST moves in the + direction to a molecule which represents a lambda term.
- Or, otherwise said, any molecule which contains the LEFT patterns of a DIST move is not one which represents a lambda term.
_______________________________________________________
3. The beta move. Reduction and evaluation.
I explain now in what sense the graphic beta move, or beta rule from chemlambda, corresponds to the beta reduction in the case of molecules which correspond to lambda terms.
Recall from part III the definition of he beta move
”
L[a1,a2,x] A[x,a4,a3] <–beta–> Arrow[a1,a3] Arrow[a4,a2]
or graphically

If we use the visual trick from the pedantic rant, we may depict the beta move as:

i.e. we use as free port variables the relative positions of the ports in the doodle. Of course, there is no node at the intersection of the two arrows, because there is no intersection of arrows at the graphical level. The chemlambda graphs are not planar graphs.”
The beta reduction in lambda calculus looks like this:
(Lx.B) C –beta reduction–> B[x:=C]
Here B and C are lambda terms and B[x:=C] denotes the term which is obtained from B after we replace all the occurrences of x in B by the term C.
I want to make clear what is the relation between the beta move and the beta reduction. Several things deserve to be mentioned.
It is of course expected that if we translate (Lx.B)C and B[x:=C] into g-patterns, then the beta move transforms the g-pattern of (Lx.B)C into the g-pattern of B[x:=C]. This is not exactly true, in fact it is true in a more detailed and interesting sense.
Before that it is worth mentioning that the beta move applies even for patterns which don’t correspond to lambda terms. Hence the beta move has a range of application greater than the beta reduction!
Indeed, look at the third figure from this post, which can’t be a pattern coming from a lambda term. Written as a g-pattern this is L[a,b,c] A[c,b,d]. We can apply the beta move and it gives:
L[a,b,c] A[c,b,d] <-beta-> Arrow[a,d] Arrow[b,b]
which can be followed by a COMB move
Arrow[a,d] Arrow[b,b] <-comb-> Arrow[a,d] loop
Graphically it looks like that.

In particular this explains the need to have the loop and Arrow graphical elements.
In chemlambda we make no effort to stay inside the collection of graphs which represent lambda terms. This is very important!
Another reason for this is related to the fact that we can’t check if a pattern comes from a lambda term in a local way, in the sense that there is no local (i.e. involving an a priori bound on the number of graphical elements used) criterion which describes the patterns coming from lambda terms. This is obvious from the previous observation that FOTREEs connect to the syntactic tree lower than their roots.
Or, chemlambda is a purely local graph rewrite system, in the sense that the is a bound on the number of graphical elements involved in any move.
This has as consequence: there is no correct graph in chemlambda. Hence there is no correctness enforcement in the formalism. In this respect chemlambda differs from any other graph rewriting system which is used in relation to lambda calculus or more general to functional programming.
Let’s go back to the beta reduction
(Lx.B) C –beta reduction–> B[x:=C]
Translated into g-patterns the term from the LEFT looks like this:
L[a,x,b] A[b,c,d] C[c] FOTREE[x,a1,…,aN] B[a1,…,aN, a]
where
- C[c] is the translation as a g-pattern of the term C, with the out port “c”
- FOTREE[x,a1,…,aN] is the FOTREE which connects to the left.out port of the node L[a,x,b] and to the ports a1, …, aN which represent the places where the lambda term variable “x” occurs in B
- B[a1,…,aN.a] is a notation for the g-pattern of B, with the ports a1,…,aN (where the FOTREE connects) mentioned, and with the out port “a”
The beta move does not need all this context, but we need it in order to explain in what sense the beta move does what the beta reduction does.
The beta move needs only the piece L[a,x,b] A[b,c,d]. It is a local move!
Look how the beta move acts:
L[a,x,b] A[b,c,d] C[c] FOTREE[x,a1,…,aN] B[a1,…,aN, a]
<-beta->
Arrow[a,d] Arrow[c,x] FOTREE[x,a1,…,aN] B[a1,…,aN, a]
and then 2 comb moves:
Arrow[a,d] Arrow[c,x] C[c] FOTREE[x,a1,…,aN] B[a1,…,aN, a]
<-2 comb->
C[x] FOTREE[x,a1,…,aN] B[a1,…,aN,d]
Graphically this is:

The graphic beta move, as it looks on syntactic trees of lambda terms, has been discovered in
Wadsworth, Christopher P. (1971). Semantics and Pragmatics of the Lambda Calculus. PhD thesis, Oxford University
This work is the origin of the lazy, or call-by-need evaluation in lambda calculus!
Indeed, the result of the beta move is not B[x:=C] because in the reduction step is not performed any substitution x:=C.
In the lambda calculus world, as it is well known, one has to supplement the lambda calculus with an evaluation strategy. The call-by-need evaluation explains how to do in an optimized way the substitution x:=C in B.
From the chemlambda point of view on lambda calculus, a very interesting thing happens. The g-pattern obtained after the beta move (and obvious comb moves) is
C[x] FOTREE[x,a1,…,aN] B[a1,…,aN,d]
or graphically

As you can see this is not a g-pattern which corresponds to a lambda term. That is because it has a FOTREE in the middle of it!
Thus the beta move applied to a g-pattern which represents a lambda term gives a g-patterns which can’t represent a lambda term.
The g-pattern which represents the lambda term B[x:=C] is
C[a1] …. C[aN] B[a1,…,aN,d]
or graphically

In graphic lambda calculus, or GLC, which is the parent of chemlambda we pass from the graph which correspond to the g-pattern
C[x] FOTREE[x,a1,…,aN] B[a1,…,aN,d]
to the g-pattern of B[x:=C]
C[a1] …. C[aN] B[a1,…,aN,d]
by a GLOBAL FAN-OUT move, i.e. a graph rewrite which looks like that
if C[x] is a g-pattern with no other free ports than “x” then
C[x] FOTREE[x, a1, …, aN]
<-GLOBAL FAN-OUT->
C[a1] …. C[aN]
As you can see this is not a local move, because there is no a priori bound on the number of graphical elements involved in the move.
That is why I invented chemlambda, which has only local moves!
The evaluation strategy needed in lambda calculus to know when and how to do the substitution x:C in B is replaced in chemlambda by SELF-MULTIPLICATION.
Indeed, this is because the g-pattern
C[x] FOTREE[x,a1,…,aN] B[a1,…,aN,d]
surely has places where we can apply DIST moves (and perhaps later FAN-IN moves).
That is for the next post.
___________________________________________________

![H[a\rightarrow b]](https://s0.wp.com/latex.php?latex=H%5Ba%5Crightarrow+b%5D&bg=ffffff&fg=2b2b2b&s=0&c=20201002)





 corresponds to
corresponds to 
 rewrites to
rewrites to  and
and 
 rewrites to
rewrites to 


 , of
, of 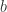 and
and  .
. 

 and
and 



 and
and 
 and
and 
 and
and  reduce to the same term
reduce to the same term 
 and
and  reduce to the same expression
reduce to the same expression 


 CO-COMM and CO-ASSOC rewrites for the FO (fanout) which are, due to the orientation of the edges, really like graphical, AST forms of co-commutativity and co-associativity
CO-COMM and CO-ASSOC rewrites for the FO (fanout) which are, due to the orientation of the edges, really like graphical, AST forms of co-commutativity and co-associativity






























 and for any
and for any  zipper combinators, the zipper graph obtained by connecting the out arrows of the zipper combinators to the in arrows of the
zipper combinators, the zipper graph obtained by connecting the out arrows of the zipper combinators to the in arrows of the  half-zipper is a zipper combinator.
half-zipper is a zipper combinator.
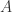 , there is a number
, there is a number  and a succession of
and a succession of 

 zipper combinators.
zipper combinators.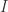 combinator, it happens like this:
combinator, it happens like this:
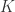 is described next:
is described next:
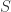 is absorbed by the following succession of moves:
is absorbed by the following succession of moves:







